


Section: New Results
Molecular Docking
Development of a new Knowledge-Based Potential for Protein-Ligand Interactions
Participants : Sergei Grudinin, Georgy Cheremovskiy.
Macromolecular complexes formed by proteins with small molecules (ligands) play an important role in many biological processes such as signal transduction, cell regulation, etc. Experimental methods for determining the structures of molecular complexes have a very high cost and still involve many difficulties. Therefore, computational methods, such as molecular docking, are typically used for predicting binding modes and affinities, which are essential to understand molecular interaction mechanisms and design new drugs.
Databases containing three-dimensional protein-ligand structures determined by experimental techniques grow very rapidly. In 2011, the PDB (Protein Data Bank) contained about 70,000 of protein structures, with almost 8,000 structures of protein-ligand complexes having refined binding affinity data. The CSD (Cambridge Structural Database), a database for small molecules, contained about 500,000 entries at the beginning of 2012. Thus, we believe that computational tools based on statistical information extracted from three-dimensional structures of protein-ligand complexes will play an ever more increasing role in the functional study of proteins as well as in structure-based drug design and other fields.
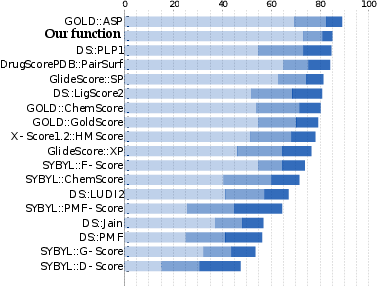
We proposed and validated a new statistical method that predicts binding modes and affinities of protein-ligand complexes. To do so, we have developed a novel machine-learning-based approach. Precisely, we have formulated a new optimization problem with 30,000 unknowns, whose solution is a scoring function. We trained the scoring function on 6,000 structures of protein-ligand complexes of high accuracy from the PDB database. Despite the very high dimensionality of the optimization problem, we manage to solve it on a desktop computer in just a few hours.
Our scoring function has three major applications in drug-design:
Docking: determination of the binding site of a ligand bound to a protein.
Ranking: identifying a set of ligands with the highest binding affinity for the given protein target by screening a large ligand database.
Binding constants prediction: prediction of the absolute value of the binding constant of a protein-ligand complex.
The success rates of our method rank it among the top three methods currently available. Thus, we believe that our scoring function is the first one that performs well in all three major applications in drug-design.
|
DockTrina
Participants : Sergei Grudinin, Petr Popov.
We derived analytical formulas for fast evaluation of the Root-Mean-Square-Deviation (RMSD) between rigid protein structures. This work resulted in a RMSD library containing algorithms to calculate the RMSD between two proteins in constant time. Based on this library we introduced an efficient algorithm to predict triangular protein structures and implemented it into the DockTrina software. We collected bound benchmarks of 220 protein trimers with and without symmetry properties from the Protein Data Bank and demonstrated the superiority of DockTrina over standard combinatorial algorithms aimed at predicting nonsymmetrical protein trimers.
Machine Learning for Structural Biology
Participants : Sergei Grudinin, Petr Popov, Mathias Louboutin.
We developed a new formulation of the machine learning optimization problem to predict protein–protein interactions. We implemented several optimization strategies, both in dual and primal. We studied the effect of different types of loss-functions on the quality of the prediction. We also tested the efficiency of three descent algorithms, Nesterov descent, gradient descent, and stochastic descent. We demonstrated that generally, primal optimization is faster compared to dual optimization. In the primal, Nesterov descent has a better convergence compared to the gradient descent. Finally, stochastic algorithms often provide a better convergence compared to deterministic algorithms. All the studied algorithms were implemented as a stand-alone library.